Edit Content




According to Longboard and its websites, Developmental and Epileptic Encephalopathy (DEE) syndromes (DEEs) refer to a group of severe heterogeneous epilepsies that are characterized by drug resistant seizures and significant developmental delay like Lennox Gastaut Syndrome. Importantly, if seizure control can be improved, developmental delay may slow. Most DEEs begin early in life, often starting in infancy. Children can have frequent and severe seizures which may be of multiple types. Epileptic spasms, tonic or atonic seizures and myoclonic seizures, among other seizure types, can be seen. In many cases, seizures are life long, although in some instances they can abate with time with certain syndromes or specific causes
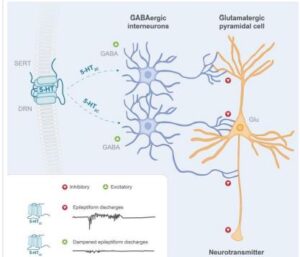
LP352 is a highly selective, oral, centrally acting, next- generation 5-HT2c receptor superagonist in development for the potential treatment of seizures associated with developmental and epileptic encephalopathies (DEEs) such as Dravet syndrome, Lennox-Gastaut syndrome (LGS), tuberous sclerosis complex (TSC), CDKL5 deficiency disorder (CDD), and other epileptic disorders.
LP352 is designed to modulate GABA inhibition and, as a result, suppress the central hyperexcitability that is characteristic of seizures. LP352 has demonstrated negligible observed impact on 5-HT2b and 5-HT2a receptor subtypes in the Company’s preclinical studies to date. 5-HT2b and 5-HT2a receptor agonism have been associated with significant adverse effects. LP352 has novel chemistry and attributes, and was designed to be more specific and selective for the 5-HT2c receptor subtype, giving it the potential to reduce
seizures in DEE patients while overcoming the known or perceived safety limitations of available drugs in the 5-HT2 class. LP352 is currently being evaluated in the Phase 1b/2a PACIFIC Study in approximately 50 participants with a range of DEEs.
The PACIFIC Study is a Phase 1b/2a clinical study evaluating adult participants with DEEs.
An individual may qualify for the study if they:
More information: info@HawaiiNeuroscience.com or Epilepsy Research Unit Hotline (808) 564-6141 or Comprehensive Epilepsy Center at Hawaii Pacific Neuroscience (808) 261-4476.
According to Sage Therapeutic website, Huntington’s disease (HD) is a rare, inherited neurodegenerative disease that progresses over time. Up to 30,000 adults are diagnosed with HD in the U.S. each year. Symptoms usually appear between ages 30–45, worsen over the following 15–20 years, and ultimately lead to death. Psychiatric and cognitive symptoms can severely affect people with HD. “HD is an autosomal dominant genetic disorder that impacts the brain and by nature numerous generations of a family. Cognitive decline is often one of the earliest signs of the disease and this decline, in addition to other symptoms, results in a devastating impact on independence, general functioning, and quality of life.
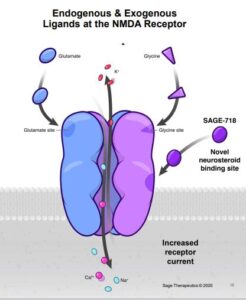
SAGE-718, Sage’s first-in-class NMDA receptor PAM and lead neuropsychiatric drug candidate, is in development as a potential oral therapy for cognitive disorders associated with NMDA receptor dysfunction, potentially including Huntington’s disease (HD), Parkinson’s disease (PD) and Alzheimer’s disease (AD). Ongoing studies aim to evaluate whether SAGE-718 may have the potential to improve cognitive symptoms for these difficult-to-treat disorders.
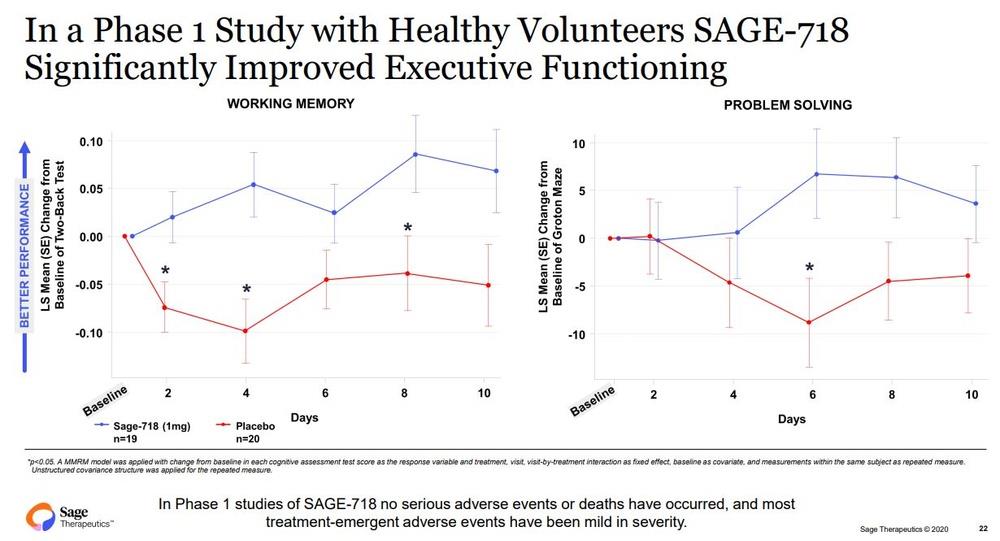
In studies to date, treatment with SAGE-718 has been associated with improved cognitive performance, particularly in the domain of executive functioning. The FDA Fast Track Designation is an important milestone in the development of SAGE-718, as it provides opportunities to engage collaboratively with the FDA to further clinical development and future regulatory review of SAGE-718 for the treatment of HD
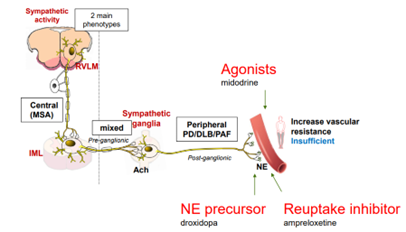 According to Therevance MSA is a progressive brain disorder that affects movement and balance and disrupts the function of the autonomic nervous system. The autonomic nervous system controls body functions that are mostly involuntary. One of the most frequent autonomic symptoms associated with MSA is a sudden drop in blood pressure upon standing (nOH).1 There are approximately 50,000 MSA patients in the US2 and 70-90% of MSA patients experience nOH symptoms.3 Despite available therapies, many MSA patients remain symptomatic with nOH.
According to Therevance MSA is a progressive brain disorder that affects movement and balance and disrupts the function of the autonomic nervous system. The autonomic nervous system controls body functions that are mostly involuntary. One of the most frequent autonomic symptoms associated with MSA is a sudden drop in blood pressure upon standing (nOH).1 There are approximately 50,000 MSA patients in the US2 and 70-90% of MSA patients experience nOH symptoms.3 Despite available therapies, many MSA patients remain symptomatic with nOH.
Neurogenic orthostatic hypotension (nOH) is a rare disorder defined as a fall in systolic blood pressure of >20 mm Hg or diastolic blood pressure of >10 mm Hg, within 3 minutes of standing. Severely affected patients are unable to stand for more than a few seconds because of their decrease in blood pressure, leading to cerebral hypoperfusion and syncope. A debilitating condition, nOH results in a range of symptoms including dizziness, lightheadedness, fainting, fatigue, blurry vision, weakness, trouble concentrating, and head and neck pain.
Ampreloxetine (TD-9855) is an investigational, once-daily norepinephrine reuptake inhibitor in development for the treatment of symptomatic nOH in patients with multiple system atrophy (MSA). Patients with MSA may benefit from ampreloxetine treatment due to the presence of central autonomic pathway degeneration and intact peripheral postganglionic fibers that is specific to MSA. As a NET re-uptake inhibitor, ampreloxetine may enhance the function of the residual sympathetic nerves resulting in increases in norepinephrine levels, standing BP, and reduction in symptoms of nOH in patients with MSA.
2022 November data presented at the American Autonomic Society 33rd International Symposium presented Phase 3 results (Study 0170) showed a benefit to MSA patients in the study that was observed in multiple endpoints including Orthostatic Hypotension Symptom Assessment (OHSA) composite, Orthostatic Hypotension Daily Activities Scale (OHDAS) composite, Orthostatic Hypotension Questionnaire (OHQ) composite and OHSA #1.
The CYPRESS Study is a Phase 3, Multi-center, Randomized Withdrawal and Long Term Extension Study of Ampreloxetine for the Treatment of Symptomatic Neurogenic Orthostatic Hypotension:
More information: info@HawaiiNeuroscience.com or Clinical Research Center Hotline (808) 564-6141 or Center for Rare Neurological Diseases at Hawaii Pacific Neuroscience (808) 261-4476.















